Syndrome de Bloom: symptômes, causes et traitement
Le syndrome de Bloom (BS) est une maladie rare de la transmission autosomique récessive qui se caractérise principalement par trois aspects: retard de croissance, hypersensibilité au soleil et télangiectasie du visage (dilatation des vaisseaux capillaires). Ces patients présentent une instabilité génomique qui les prédispose à développer facilement un cancer.
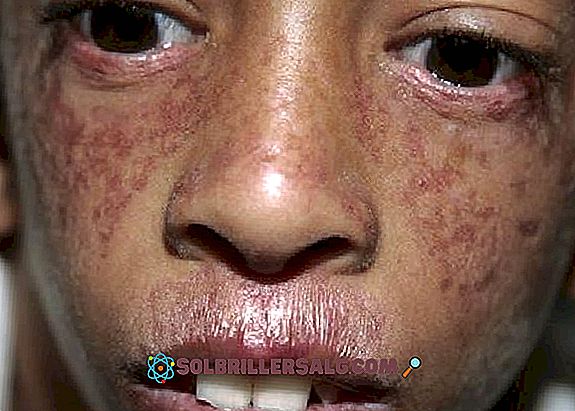
Il a été découvert par le dermatologue David Bloom en 1954 grâce à l'observation de plusieurs patients présentant un nanisme et un érythème télangiectatique (peau rougie en raison de la dilatation des capillaires sanguins) (Elbendary, 2015).
Ce syndrome peut également être appelé érythème télangiectasique congénital ou syndrome de Bloom-Torre-Machacek.
Causes du syndrome de Bloom
Le syndrome de Bloom est une maladie autosomique récessive, c'est-à-dire qu'une mutation doit se produire dans les deux allèles du gène BLM, à la fois de la mère et du père (Ellis et al., 1995). Les parents ne doivent pas nécessairement être atteints de cette maladie, mais ils peuvent être porteurs du gène muté sans présenter de symptômes.
Plus de 60 mutations ont été trouvées dans le gène BLM dans le syndrome de Bloom, les plus fréquentes étant la délétion de 6 nucléotides en position 2281 et la substitution par 7 autres (Elbendary, 2015).
Selon Genetics Home Reference (2016), le gène BLM est responsable de l'envoi des instructions pour la création de la protéine RecQ, qui fait partie de la famille des hélicases.
Ce que font les hélicases, c'est joindre l'ADN et en séparer temporairement les deux brins, généralement liés en spirale, dans le but de développer des processus tels que la réplication (ou copie de l'ADN), la préparation à la division et à la réparation cellulaires. des dommages à l'ADN.
En bref, les hélicases RecQ sont importantes pour maintenir la structure de l'ADN et sont donc connues sous le nom de "soigneurs du génome".
Par exemple, lorsqu'une cellule va se diviser pour former deux nouvelles cellules, l'ADN contenu dans les chromosomes doit être copié de manière à ce que chaque nouvelle cellule possède deux copies de chaque chromosome: une du père et une autre de la mère.
L'ADN copié de chaque chromosome a deux structures identiques, appelées chromatides soeurs, et elles sont liées au début, avant la division des cellules.
À ce stade, ils échangent des fragments d’ADN entre eux; ce qu'on appelle l'échange de chromatides soeurs. Il semble que ce processus soit altéré dans la maladie de Bloom, car la protéine BLM est endommagée et c'est ce qui contrôle les échanges appropriés entre les chromatides sœurs et que l'ADN reste stable au moment de la copie.
En fait, il existe en moyenne 10 échanges de plus que la normale entre chromatides dans le syndrome de Bloom (Seki et al., 2006).
D'autre part, il existe également des ruptures dans le matériel génétique de cette maladie, qui entraînent une détérioration des activités cellulaires normales qui, en raison de l'absence de la protéine BLM, ne peuvent pas être réparées.
En fait, certains experts qualifient ce syndrome de "syndrome de rupture chromosomique", car il est lié à un grand nombre de ruptures et de réarrangements des chromosomes.
Cette instabilité des chromosomes augmente la probabilité de développer des maladies. Par exemple, en raison de l’absence de protéine BLM, ils ne peuvent pas récupérer des dommages de l’ADN pouvant provoquer une lumière ultraviolette et, par conséquent, ces patients sont photosensibles.
En outre, les personnes atteintes présentent un déficit immunitaire qui les rend plus vulnérables aux infections.
Par contre, ils ont une forte probabilité de développer un cancer dans n'importe quel organe en raison d'une division incontrôlée des cellules, apparaissant principalement comme une leucémie (type de cancer du sang caractérisé par un excès de globules blancs) et par un lymphome (cancer du ganglion lymphatique du système). immunisé).
Des défaillances ont également été constatées dans l’action du gène FANCM, responsable du codage des protéines MM1 et MM2, qui servent également à réparer les dommages de l’ADN.
Ce sont ceux qui ont été liés à la fois à ce syndrome et à l'anémie de Fanconi. C'est pourquoi on voit que ces deux maladies sont similaires dans leur phénotype et dans leur prédisposition aux tumeurs hématologiques et à une insuffisance de la moelle osseuse.
Quoi qu'il en soit, les mécanismes moléculaires qui affectent les chromosomes dans le syndrome de Bloom sont toujours à l'étude.
Quelle est sa prévalence?
Le syndrome de Bloom est relativement peu fréquent, seuls environ 300 cas décrits dans la littérature médicale sont connus. Bien que cette maladie touche de nombreux groupes ethniques, elle semble être beaucoup plus fréquente chez les Juifs ashkénazes, représentant 25% des patients atteints de ce syndrome.
En fait, au sein de ce groupe ethnique, la fréquence de présentation du syndrome peut atteindre 1%. Il a également été trouvé, bien que moins fréquemment, dans des familles japonaises.
En ce qui concerne le sexe, les hommes semblent être un peu plus susceptibles d'avoir la maladie que les femmes, la proportion étant de 1, 3 homme pour 1 femme.
Quels sont ses symptômes?
Cette maladie survient déjà au cours des premiers mois de la vie et, pour le moment, aucun patient n'a vécu plus de 50 ans.
- Les tumeurs malignes : causées par l'instabilité génomique, comme expliqué ci-dessus, sont la principale cause de décès chez les personnes atteintes de ce syndrome. Selon l'Organisation nationale pour les maladies rares (2014), environ 20% des personnes atteintes du syndrome de Bloom développeront un cancer. Ces patients ont entre 150 et 300 fois plus de risques de développer un cancer que les personnes ne présentant pas ce trouble.
- Déficit immunitaire de sévérité variable selon les patients et prédisposant à diverses infections. Cela est dû à des déficits dans la prolifération des lymphocytes (globules blancs), à des problèmes de synthèse d'immunoglobuline (anticorps du système immunitaire) et à une faible réponse à la stimulation par des mitogènes (qui contrôlent la division et la croissance des cellules).
- Les défauts dans les lymphocytes T et B sont fréquents, ce qui affecte le développement du système immunitaire.
- Un dysfonctionnement du système immunitaire peut entraîner une infection de l'oreille (principalement une otite moyenne), une pneumonie ou d'autres signes tels que diarrhée et vomissements.
- Photosensibilité : c'est une sensibilité excessive de l'ADN aux rayons ultraviolets, qui s'endommage. Il est considéré comme une forme de phototoxicité ou de mort cellulaire qui nuit à la peau de la personne touchée lorsque le soleil frappe.
- réduction de la fertilité ou de l'infertilité . En fait, chez les hommes, il est impossible d’attendre. Chez les femmes, il y a une ménopause très précoce.
- manifestations cutanées : en plus de la photosensibilité, il se produit également une poikilodermie, une altération de la peau qui se produit principalement dans le cou, des zones apparentes hypopigmentées, d'autres zones hyperpigmentées, des télangiectasies et une atrophie. On observe couramment des taches rouges sur la peau associées à l'exposition au soleil (surtout au visage).
- Un autre problème cutané observé est la télangiectasie, qui se manifeste par des éruptions rougeâtres au visage provoquées par la dilatation de petits vaisseaux sanguins. Il apparaît comme un motif "papillon" couvrant le nez et les joues.
- Des taches brunes ou grises anormales peuvent également apparaître sur d'autres parties du corps (taches "café au lait").
- Retard dans le développement qui se manifeste déjà chez les bébés. Les plus petits ont généralement une tête et un visage distinctifs, plus étroits et plus petits que la normale.
- Environ 10% des personnes concernées finissent par développer un diabète (Organisation nationale pour les maladies rares, 2014).
- voix très forte .
- Altérations dans les dents .
- Anomalies des yeux, des oreilles ( oreilles bien en évidence), des mains ou des pieds (comme une polydactylie, qui se produit lorsque le patient a plus de doigts que la normale).
- Kystes pilonidaux.
- Problèmes d'alimentation : ils se manifestent surtout chez les bébés et les jeunes enfants, manifestant un manque d'intérêt pour l'alimentation. Il est accompagné à plusieurs reprises de reflux gastro-oesophagien sévère.
- Les capacités intellectuelles sont variables, de sorte que chez certains patients, elles sont plus détériorées et chez d'autres, elles sont dans la normale.
Comment est-il diagnostiqué?
Il peut être diagnostiqué par l'un des tests suivants:
- Tests cytogénétiques qui mesurent les aberrations chromosomiques et le niveau d'échange des chromatides soeurs.
Il est possible d’observer la présence d’associations à quatre radiales (échange de chromatides à quatre bras) dans des lymphocytes cultivés dans le sang, afin de vérifier s’il existe de hauts niveaux d’échange de chromatides soeurs dans une cellule quelconque, dans les lacunes, les ruptures ou les réarrangements de chromatides; Ou voyez directement s'il y a des mutations dans le gène BLM.
Ces tests permettent de détecter un individu en bonne santé porteur de mutations dans le gène BLM et de les transmettre à leur progéniture.
La Food and Drug Administration des États-Unis (FDA) a annoncé en février 2015 la commercialisation par "23andMe" d'un test génétique qui pourrait s'avérer utile pour détecter rapidement la présence de cette maladie.
La présence de ce syndrome doit être suspectée si ces conditions cliniques existent:
- Retard dans la croissance significative observée depuis la période intra-utérine.
- Présence d'érythèmes sur la peau du visage après une exposition au soleil.
Ne pas confondre avec ...
Les syndromes suivants doivent être pris en compte pour les éliminer avant le diagnostic du syndrome de Bloom:
- Autres syndromes d'instabilité chromosomique récessifs autosomiques récessifs associés à des ruptures et à des réarrangements de chromosomes, rendant le sujet particulièrement vulnérable à certains types de cancer, tels que: anémie de Fanconi, ataxie télangiectasie ou xeroderma pigmentosa impliquant d'autres gènes et non le BLM .
- le syndrome de Cockayne, qui consiste en un trouble héréditaire se manifestant par un retard de développement, une photosensibilité et une apparence de vieillissement à un jeune âge. C'est une forme rare de nanisme.
- Syndrome de Rothmund-Thomson : extrêmement rare et se manifestant par des anomalies cutanées typiques, des défauts des poils, des cataractes juvéniles, une petite taille et des altérations du squelette telles que des malformations cranofaciales. Il ressemble au syndrome de Bloom dans les cas d'inflammation de la peau, de poïkilodermie, de dégénérescence cutanée (atrophie) et de télangiectasie.
Traitement
Il n'y a pas de traitement spécifique pour le syndrome de Bloom, c'est-à-dire pour le nombre excessif de mutations. Les interventions visent plutôt à atténuer les symptômes, à offrir un soutien et à prévenir les complications.
- Essayez de ne pas vous exposer directement au soleil.
- Utilisez un écran solaire adéquat.
- Suivi par un dermatologue pour traiter les taches, les rougeurs et les inflammations de la peau.
- Utilisez des antibiotiques pour les infections.
- Des examens médicaux périodiques pour détecter d'éventuels cas de cancer, principalement lorsque ces patients atteignent l'âge adulte. Nous devons essayer d'être attentifs aux symptômes possibles, car certaines tumeurs nécessitent un retrait chirurgical précoce pour pouvoir être récupérées. Certaines méthodes de diagnostic précoce du cancer sont la mammographie, la cytologie papanicolaou ou vaginale ou la coloscopie.
- Contrôler que ces enfants reçoivent les nutriments nécessaires en essayant d'intervenir dans le reflux digestif. Pour cela, un tube peut être placé dans la partie supérieure du tractus intestinal pour une alimentation complémentaire pendant votre sommeil. Cela peut augmenter un peu les dépôts de graisse, mais cela ne semble pas avoir d’effet sur la croissance elle-même.
- Examiner l'existence du diabète pour le traiter le plus tôt possible.
- Si l'individu est atteint d'un cancer, une greffe de moelle osseuse peut être envisagée.
- Groupes familiaux et autres groupes de soutien et associations ayant des maladies similaires, de sorte que la personne touchée se développe en tant que personne et jouisse d'une qualité de vie optimale.
- Si la famille a eu des cas de cette maladie ou par la famille du conjoint, un conseil génétique serait utile pour obtenir des informations sur la nature, l'héritage et les conséquences de ce type de troubles afin de contribuer à la prise de décision médicale. et personnelle.







