Maladie de Tay-Sachs: symptômes, causes et traitement
La maladie de Tay-Sachs est une pathologie du patrimoine génétique qui affecte le système nerveux central. Aussi connu sous le nom de gangliosidose GM2, il est causé par la déficience d'une enzyme essentielle appelée bêta-hexosaminidase A.
Cette enzyme est responsable de la décomposition et de l'élimination des déchets toxiques produits par l'activité cérébrale. En l'absence de l'enzyme, les résidus s'accumulent sous forme de gangliosides et provoquent la détérioration du système nerveux central.

Les dommages causés par la maladie de Tay-Sachs sur les neurones sont irréversibles et touchent principalement le cerveau et la moelle épinière. La détérioration neuronale entraîne des troubles neurologiques progressifs.
Les symptômes se caractérisent généralement par des tremblements de la main, des troubles de la parole, une faiblesse musculaire et une perte d'équilibre.
La surdité, la perte de capacité visuelle, les crises d'épilepsie, un retard de croissance, l'irritabilité, l'apathie et le retard mental sont d'autres signes typiques.
Actuellement, il n'y a pas de traitement pour guérir la maladie. Les personnes qui en souffrent meurent généralement entre 4 et 5 ans après le diagnostic. La guérison de Tay-Sachs est l’un des principaux défis de la recherche scientifique actuelle.
Caractéristiques de la maladie de Tay-Sachs
La maladie de Tay-Sachs est comprise dans Glangliosidose GM2. Il s’agit d’un groupe de maladies lysosomales dans lesquelles il existe une accumulation de gagliosides de GM2 qui ne sont pas métabolisés.
Les raisons pour lesquelles ils ne sont pas métabolisés peuvent être dus à la déficience en enzymes appelées hexosaminidase A et hexosaminidase B. Ou en raison de la déficience en protéine activante de GM2.
Actuellement, trois mutations ont été décrites dans trois gènes différents pouvant produire une glangliosidose GM2: la maladie de Tay-Sachs, la maladie de Sandhoff et le déficit en activateur de GM2.
Les pathologies du dépôt lisomal appartiennent au groupe des maladies métaboliques congénitales, parmi lesquelles environ 70 maladies sont connues. Toutes ces altérations sont caractérisées par la déficience en une enzyme vitale.
Tous n’affectent pas le cerveau, mais beaucoup le font. C'est le cas de Tay-Sachs qui est défini comme une gagliosidose à GM2 provoquée par un déficit en hexosaminidase A.
La déficience de cette enzyme est produite par des mutations dans la sous-unité alpha de ladite enzyme. Pour cette raison, Tay-Sachs est considéré comme une pathologie génétique.
Les symptômes
La maladie de Tay-Sachs présente une série de symptômes communs qui se manifestent dans tous les cas. Cependant, la symptomatologie peut se manifester de différentes manières chez chaque patient.
La variabilité de la forme clinique adoptée dépend principalement de la mutation héritée de la pathologie. De même, l'évolution de la maladie est directement liée à la quantité d'hexosaminidase que possède la personne atteinte de Tay-Sachs.
Plus la quantité d'hexosaminidase est faible, plus l'accumulation de gangliosidose est importante et, par conséquent, plus les lésions cérébrales et les symptômes présentés sont sévères. En réponse à ces critères, trois formes cliniques de Tay-Sachs ont été postulées.
Enfant Tay-Sachs
Cette variante de la maladie, également appelée Tay-Sachs du nourrisson précoce ou aigu, est la forme classique de la pathologie. De même, c'est aussi le plus agressif et le plus fulminant.
Les enfants touchés par Tay-Sachs n’ont généralement pas d’hexosaminidase. La destruction du cerveau commence donc à un stade très précoce. Le plus commun est qu'il commence déjà pendant la grossesse.
À la naissance, le bébé présente un état de santé sans aucun type de symptomatologie. Cependant, entre trois et six mois de vie, les premières manifestations commencent à apparaître.
A ce moment, il est connoté alors que le développement normal ralentit et que des problèmes de vision apparaissent. En particulier, le contact visuel et la concentration visuelle sont réduits.
Une audition accrue, provoquant une réaction de surprise exagérée à certains stimuli auditifs (hyperacousie), est un autre des principaux symptômes de ces stades.
De même, un autre symptôme pathognomonique de la maladie est la couleur rouge de la macula, une région proche du nerf optique. Cette manifestation est généralement l’un des principaux signes qui permettent le diagnostic car elle peut être détectée par un simple examen ophtalmologique.
Au fil du temps, la perte de capacités psychomotrices augmente progressivement. Il y a aussi une réduction du tonus musculaire (hypotonie) qui finit par causer une faiblesse généralisée.
Par la suite, le bébé devient incapable de rouler, de ramper, de s’asseoir et de saisir des objets. Tout comme il apparaît une incapacité à avaler et des complications respiratoires, de la spasticité et de la rigidité des extrémités.
En général, à l'âge de 2 ans, l'enfant a déjà une tétraplégie spastique, des crises d'épilepsie et des crises récurrentes. La mobilité musculaire, la vision et la plupart des capacités mentales sont complètement perdues.
Dans la plupart des cas, il existe une augmentation marquée de la taille du crâne et des dommages importants au système nerveux. La mortalité de cette forme clinique de la maladie de Tay-Sachs survient généralement entre 2 et 4 ans.
Tay-Sachs juvénile
Contrairement au nouveau-né Tay-Sachs, les sujets atteints ne sont pas nés avec l'absence totale d'hexosaminidase. Dans ce cas, la production de cet enzyme chez les bébés est généralement faible, car elle se désintègre au cours des premières années de la vie.
De cette manière, la symptomatologie est généralement un peu plus tardive et ne présente habituellement pas de manifestations avant 2 à 5 ans environ. L'établissement de l'âge d'apparition de cette forme clinique de Tay-Sachs soulève toutefois certaines controverses.
Certains auteurs postulent que cela commence entre la première et la dixième année de vie, tandis qu'un autre convient entre 2 et 18 ans. Cependant, bien que les symptômes apparaissent généralement tardivement, ils apparaissent rarement après l’adolescence.
La symptomatologie présentée est très similaire à celle que nous avons commentée à propos du nouveau-né Tay-Sachs. Mais le développement peut être plus lent, surtout dans les cas où les manifestations apparaissent après 5 ans de vie.
La survie de cette forme clinique est également plus variable. La majorité des personnes touchées meurent généralement entre 2 et 4 ans après le diagnostic de la maladie. Cependant, dans certains cas, la première et même la deuxième décennie de la vie peuvent être surmontées.
Tay-Sachas tard
La maladie de Tay-Sachs peut également faire ses débuts à l'âge adulte. Dans ces cas, la symptomatologie présentée et l'âge d'apparition peuvent être très variables.
En général, les premiers symptômes apparaissent pendant l'adolescence, présentant une dysarthrie, une ataxie, des tremblements et une hypotonie. Les crampes et les spasmes musculaires sont également des symptômes courants au cours des premiers stades.
Dans chaque cas, des symptômes différents peuvent apparaître, mais la faiblesse des muscles proximaux apparaît dans chacun d'entre eux. Les problèmes d'assise, de lever du lit ou de perte d'équilibre sont généralement des manifestations typiques.
Des épisodes dépressifs, des épidémies psychotiques et d'autres modifications psychologiques apparaissent jusqu'à 30% des cas de Tay-Sachs tardif. L'âge de décès de cette forme clinique de la maladie peut être très variable, mais il dépasse rarement la quatrième décennie de la vie.
Diagnostic
Afin de diagnostiquer la maladie de Tay-Sachs, les niveaux d’hexosaminidase doivent être analysés. De cette manière, l'évaluation de la symptomatologie manifestée n'est pas suffisante pour son diagnostic et une analyse biochimique est nécessaire.
Chez les nourrissons Tay-Sachs, les bébés ne présentent pas d’hexosaminidase et chez les juvéniles et les derniers Tay-Sachs, de très faibles concentrations de cette enzyme sont présentes dans le sang.
En ce sens, procéder à une analyse génétique pour confirmer la maladie et identifier les mutations du gène HEX-A qui causent un déficit en hexosaminidase est un outil très utile pour le diagnostic de la pathologie.
Enfin, les porteurs de la pathologie, les géniteurs, peuvent effectuer une analyse pour mesurer leur niveau d’hexosaminidase dans le sang. Ce dernier test est généralement conseillé pour l’étendre au groupe familial et rechercher d’autres transporteurs Tay-Sachs possibles.
Les causes
La cause de cette pathologie est une mutation d'un gène, le gène HEX-A. Ce gène se trouve sur le bras long du chromosome 15 et une mutation en provoque la maladie de Tay-Sachs.
Le gène HEX-A contient les instructions pour générer une partie très importante de l'enzyme bêta-hexosaminidase A. Lorsque le gène HEX-A est muté, cette enzyme n'est pas générée.
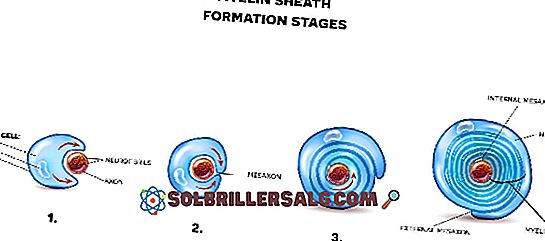
L'hexosaminidase A est située dans les lismoas, structures qui se trouvent à l'intérieur des cellules. La fonction principale de l'enzyme est de décomposer les substances toxiques dans les neurones.
Plus précisément, la bêta-hexosaminidase A est responsable de la décomposition d'une substance grasse appelée gangliosidoside GM2. Lorsque le corps n'est pas capable de produire cette enzyme (en raison d'une mutation génétique), il se produit une accumulation de toxines dans les neurones du cerveau. Ce fait produit une destruction progressive des neurones et l'apparition des symptômes de Tay-Sachs.
La transmission de cette pathologie s'effectue sous un schéma de transmission autosomique récessive. Par conséquent, pour développer la pathologie, il est nécessaire que les deux parents soient porteurs d'une copie de la mutation du gène. Si un seul l'est, l'enfant ne développera pas Tay-Sachs.
Les porteurs de la maladie peuvent avoir une production d'hexosaminidase légèrement inférieure à celle habituelle, mais aucun symptôme. De même, lorsque les deux parents sont porteurs, l’enfant peut avoir 3 possibilités:
- Si aucun des parents ne transmet la mutation génétique, le bébé naîtra en bonne santé et il est peu probable qu'il développe Tay-Sachs.
- Si un seul des parents porteurs transmet la mutation génétique au bébé, l'enfant ne développera pas la maladie mais sera porteur. Comme ses parents.
- Si les deux parents transmettent le gène muté au bébé, l'enfant sera affecté par Tay-Sachs. En fonction des mutations dont vous avez hérité, vous pouvez présenter certaines des variantes cliniques. Mais tôt ou tard, il développera Tay-Sachs.
Prévalence
L'origine de Tay-Sachs se trouve dans la population d'origine juive Ashkenzi. Son origine répond à la mutation du gène Hex-A est très répandue dans ce groupe ethnique.
Ainsi, la prévalence de Tay-Sachs est également beaucoup plus élevée chez les descendants d'origine juive ashkenzi. C'est-à-dire que cette pathologie est particulièrement répandue en Europe centrale et en Europe de l'est.
Plus précisément, on estime que la prévalence de Tay-Sachs dans cette population serait de 27%.
Comme pour le reste des groupes ethniques, les Tay-Sachs peuvent également être développés, mais leur prévalence est nettement plus faible.
Aujourd'hui, on estime que dans la population en général, une personne sur 360 000 serait touchée par Tay-Sachs et qu'une sur 250 serait porteuse de la pathologie.
Traitement
À l'heure actuelle, il n'existe aucun traitement pour guérir cette pathologie ou les maladies associées à Tay-Sachs. En fait, les enfants touchés n'ont aucune espérance de vie aujourd'hui.
Sans aucun doute, le traitement de cette pathologie est l’un des principaux défis de la science, qui initie de plus en plus de recherches visant à obtenir des médicaments pouvant guérir les maladies de Tay-Sachs.
En fait, le remède à cette pathologie serait également le remède à plus de 70 maladies du dépôt lyosomal. Les maladies de Parkinson, d'Alzheimer ou de sclérose en plaques sont les plus connues et les plus répandues.
Aujourd'hui, les personnes touchées par Tay-Sachs ne reçoivent que des traitements et des soins palliatifs. Celles-ci sont généralement communes dans d'autres maladies dégénératives ou neuromusculaires.
La stimulation précoce, la physiothérapie, l'ergothérapie, l'orthophonie, la déglutition, la physiothérapie respiratoire, l'hydrothérapie ou la stimulation musicale sont les traitements les plus utilisés.
Cependant, ces interventions ne font qu’améliorer le bien-être de la personne touchée par Tay-Sachs et ralentissent l’apparition des symptômes, mais ne guérissent pas la maladie.
D'autre part, des médicaments tels que le baclofène et le lévétiracétam, l'acide valproïque ou les benzodiazépines sont utilisés pour lutter contre les symptômes de la maladie, tels que la raideur musculaire, la spasticité et les convulsions.